Poster Presentation 21st International Conference on Biological Inorganic Chemistry 2025
Mechanistic analysis of the [FeFe]-hydrogenase maturase HydE via EPR and Native Mass Spectrometry (#535)
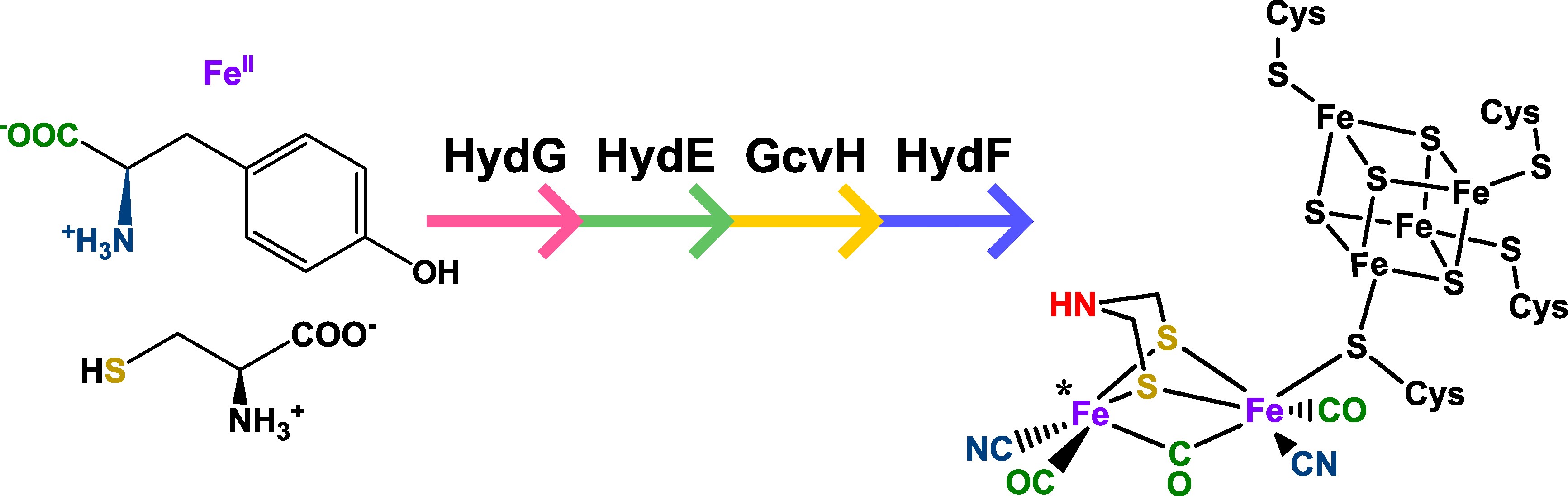
The [FeFe] Hydrogenase family of enzymes use an organometallic six-iron cofactor to interconvert protons and H2 rapidly at room temperature with minimum overpotential. This cofactor is constructed from a [Fe4S4] cluster joined via a proteinaceous cysteine to a unique organometallic [Fe2(adt)(CO)3(CN)2]2− cluster, termed "2FeH". The distal iron of this cluster is where catalysis occurs. This 2FeH cluster is synthesized from free Fe3+, cysteine, and tyrosine by the maturases HydE,F, and G with the assistance of the glycine cleavage system (Figure 1). The mechanism of HydG is known, and initial steps of the mechanism of HydE are understood, but the final product and method of product release from HydE are not known. The mechanism of HydF is also debated.
This work focuses on completing our understanding the mechanism of HydE by combining established organometallic semisynthesis methods with electron paramagnetic resonance (EPR) and native mass spectrometry (NMS). Using non-denaturing NMS of proteins allows the analysis of substrate binding, turnover, and product release in real time, as well as characterizing binding affinity and protein conformation as a function of the substrates. EPR provides complimentary information in the form of detailed electronic information at the active site.
We show here that all steps from the cleavage of the cysteinate ligand to dimerization to form the dithiolate dimer occur at HydE, raising questions about the function of HydF, which was previously assumed to be responsible for dimerization of the mononuclear precursor Complex B. This agrees with a recent computational study conducted with Prof. Lee-Ping Wang, which indicated condensation of HydE intermediates to the Fe(I) dimer. This also opens the system for further structural and biochemical analysis, and inspires speculation on the interaction modes between the Hyd maturation and glycine cleavage systems.