Poster Presentation 21st International Conference on Biological Inorganic Chemistry 2025
The Role of Structure and pKa in the Stabilization Effect of NH–S Hydrogen Bonds in Synthetic Cubane-Type Iron-Sulfur and Iron-Selenium Clusters (#516)
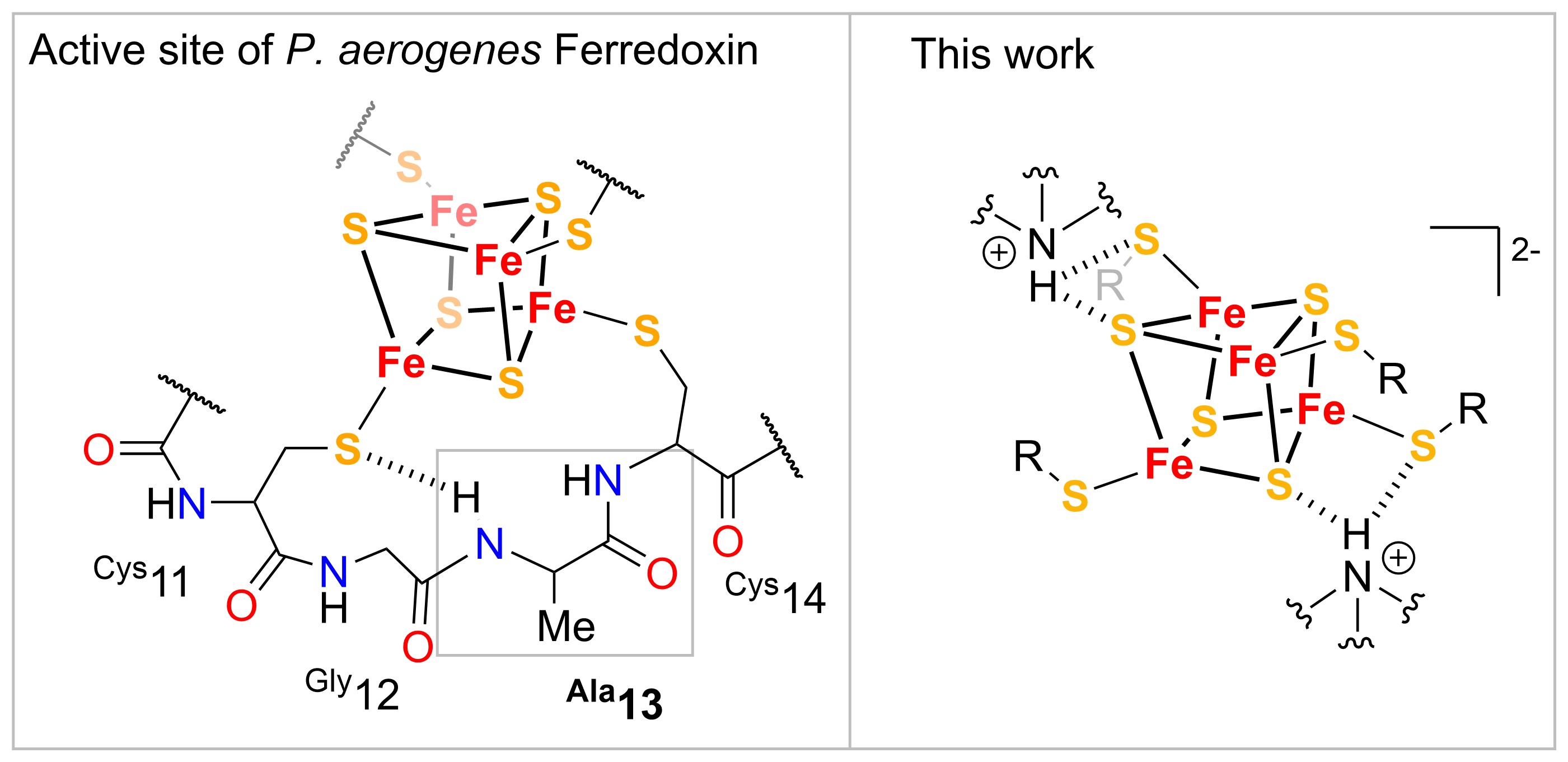
Iron sulfur (FeS) cofactors are integral components in a variety of metalloenzymes and play a central role in many biological processes due to their structural and chemical versatility as well as their unique electrochemical properties.1,2 In this context, iron-sulfur cubanes (Fe4S4) have been developed as synthetic models of Fe4S4-containing metalloenzymes, most notably high potential iron-sulfur proteins (HiPIP) and bacterial ferrodoxines, which respectively act as competent electron transfer mediators in metabolic processes.3,4 The redox properties of Fe4S4 clusters in these complex enzymatic systems is tailored by directional NH-S hydrogen bonding interactions between nitrogen atoms of polypeptide functionalities and sulfur atoms of both the Fe4S4 core and cysteine ligands as well as electrostatic interactions.5
Early work of Nakamura has demonstrated that the spatial proximity of H-bonding donor functionalities in the ligand framework of synthetic Fe4S4 clusters enables the modification of the redox potential E via NH-S hydrogen bonds in analogy to natural systems.6 Previous work in our group has focused on the dynamic modulation of the redox potential E of Fe4S4 mimics, exploiting a gated electron transfer (ET) mechanism, by encapsulation of K+ ions in the second coordination sphere close the Fe4S4 core, enabling the stabilization of highly reducing species.7,8 Inspired by this, our group has recently established the rational control of the redox potential of synthetic Fe4X4 (X= S, Se) clusters by NH-S hydrogen bonding interaction between nitrogen atoms of ammonium cations with S or Se atoms of the Fe4X4 (X= S, Se) cubane core and thiolate ligands respectively. This has been extensively studied among a systematic series of acids containing varying numbers of NH hydrogen-bond donor functionalities encompassing varying pKa strengths. The assessment of this stabilization-effect has been conducted by spectroscopic- as well as electrochemical-techniques concomitant with the isolation of solid-state molecular structure of Fe4X4 acid-base adducts. Lastly we have been able to demonstrate that NH-S hydrogen bonding enables the stabilization of reduced [Fe4X4]1+ acid-adduct complexes which are competent in proton-coupled electron transfer (PCET) reactivity to model substrates.
- 1 Helmut Beinert, Richard H. Holm, Eckard Münck, Science, 1997, 277, 653-659.
- 2 Deborah C. Johnson, Dennis R. Dean, Archer D. Smith, Michael K. Johnson, Annual Review of Biochemistry, 2005, 74,247-281.
- 3 L. C. Sieker, Elinor Adman, L. H. Jensen, Nature, 1972, 235, 40-42.
- 4 Charles W. Carter, Joseph Kraut, Stephan T. Freer, Nguyen-huu Xuong, Richard A. Alden, Robert G. Bartsch, Journal of Biological Chemistry, 1974, 249, 4212-4225.
- 5 Elinor Adamn, Keit D. Watenpauhg, Lyle H. Jensen, Proc. Nat. Acad. Sci., 1975, 72, 4854-4858.
- 6 Liam Grunwald, Martin Clémancey, Daniel Klose, Lionel Dubois, Serge Gambarelli, Gunnar Jeschke, Michael Wörle, Geneviève Blondin, Victor Mougel, PNAS, 2022, 119, 1-9.
- 7 Liam Grunwald, Mariko Inoue, Paula Cendoya Carril, Michael Wörle, Victor Mougel, Chem, 10, 365-387.
- 8 Norkazu Ueyama, Yusuke Yamada, Taka-aki Okamura, Shuuji Kimura, Akira Nakamura, Inorg. Chem., 35, 6473-6484.