Poster Presentation 21st International Conference on Biological Inorganic Chemistry 2025
Rate-Tunable Metal-Mediated Amide Bond Cleavage for the Controlled Release of Metallodrugs (#432)
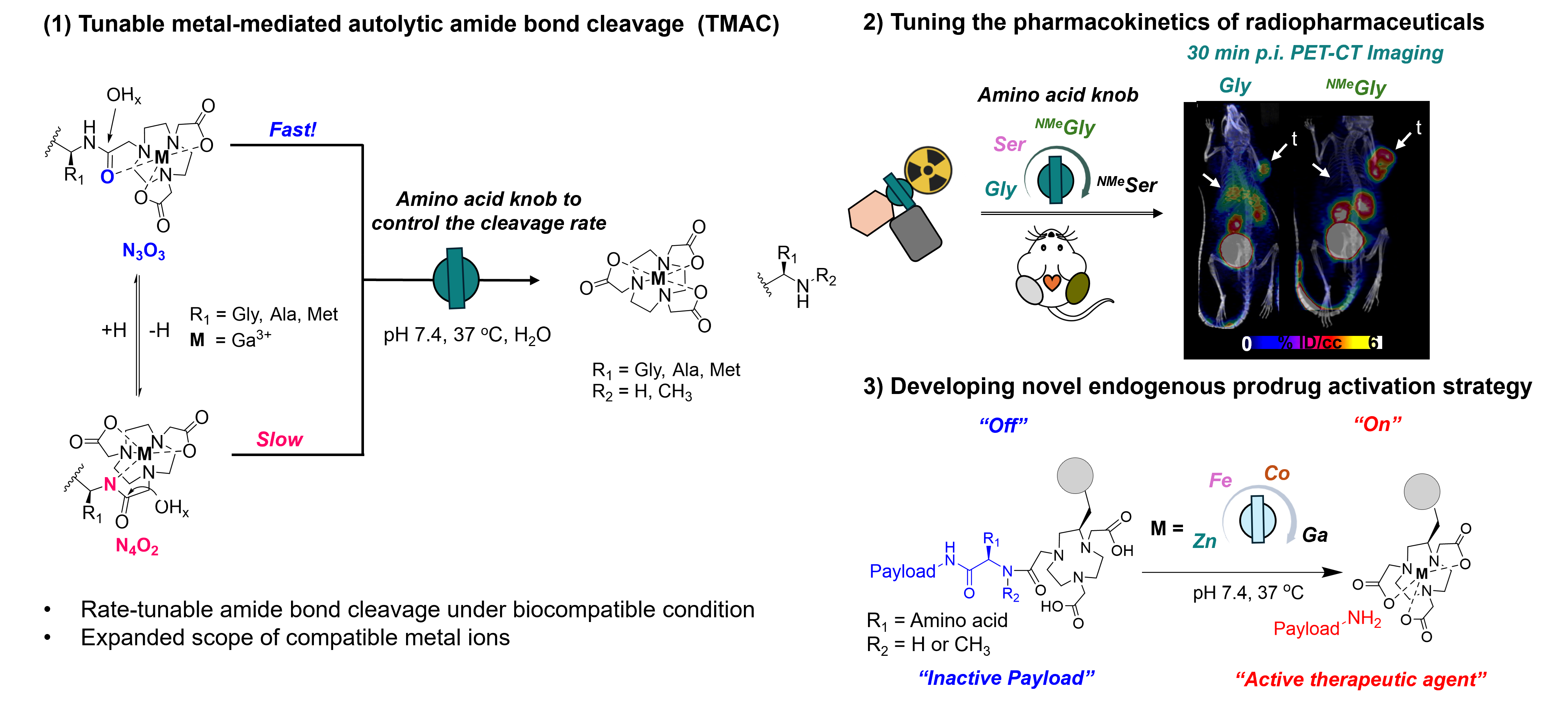
To achieve spatiotemporally controlled drug release in target tissues, prodrug activation approaches rely on exogeneous triggers such as enzymes, light irradiation, or reducing agents.1 However, their clinical application is constrained by suboptimal tissue accumulation, low catalytic turnover, and poor performance in the heterogeneous tumor microenvironment.2,3 Here, we report a novel endogenous prodrug activation strategy that the complexation of trivalent Lewis acidic metal ions selectively triggers tunable, metal-catalyzed, autolytic amide bond cleavage (TMAC) to release therapeutic agents and radiopharmaceuticals under physiological conditions. Complex-adjacent amino acids act as key modulator of cleavage kinetics, thereby optimizing the biological half-life of radiopharmaceuticals and prodrug activation in vivo. We demonstrate that trivalent metal ions, chelated by (7-amido)-1,4,7-triazonane-1,4-diyl)diacetic acid, polarize the amide bond proximal to canonical and non-canonical amino acids, forming two coordination isomers, N3O3 and N4O2, in aqueous solution (Figure 1). Subsequent hydrolysis of the polarized amide bond is facilitated by exogenous hydroxide ions and proceeds with variable rates, depending on the amino acid sidechain and N-substituent on amide. The applicability of TMAC was validated in vivo. Compounds [68Ga]Ga-23 and [68Ga]Ga-24, incorporating N-methyl-serine or N-methyl-glycine as chelate-adjacent, cleavable amino acids between the radiochelate and targeting vector, efficiently release the chelate in vivo, minimizing radiotracer accumulation in blood and liver compartments. In contrast, [68Ga]Ga-20, incorporating a non-cleavable glycine linker, exhibited elevated blood and liver uptake (Figure 2). We further validate TMAC-based prodrug release with a clinically established tubulin polymerization inhibitor, monomethyl auristatin E, which contains a terminal N-methyl-valine residue essential for its activity. Here, prodrug potency can be tuned by varying trivalent metal ions, which exhibit diverse TMAC kinetics. Ga-PD1, exhibiting rapid TMAC activation kinetics, demonstrated enhanced cytotoxic potency in cell viability assay compared to Fe-PD1, which displayed slower activation kinetics, and Zn-PD1, which was inactive (Figure 3). Collectively, TMAC represents an effective, emerging strategy for targeted prodrug release in vivo.
- (1) Liang, J.; Wei, F.; Chao, H. Smart Molecules. 2024, 2, e20240030.
- (2) Guo, Z.; Hong, H.; Zheng, Y.; Wang, Z.; Ding, Z.; Fu, Q.; Liu Z. Angew. Chem. 2022, 61, e202205014.
- (3) Jacobsen, F. E.; Lewis, J. A.; Cohen S. M. J. Am. Chem. Soc. 2006, 128, 3156-3157.